
I will keep an updated list of posts in this series:
Category Archives: Genome size
What would I do with more research support? Part Two: "Targeted exploration".
In the first post in this series, I introduced the background topic of my research focus, namely the evolution and impacts of genome size diversity in animals. Before moving on to the specific projects that I would most like to do in the near term if I had the funds, I want to discuss the basic philosophical approach that much of my lab’s work follows.
As I noted recently, there is a strong tendency among many biologists to assume that only “hypothesis-driven” science is valid and informative. I disagree with this position very strongly, as I think it causes people to focus on narrow questions and runs a real risk of making most science little more than an exercise in confirming and refining what we already know. Moreover, it is only feasible to structure one’s research in the simple, falsificationist hypothesis-testing format if there is extensive background knowledge available. When working in a new area where little is known, this is not possible.
Does this mean we should be allowed to just stumble around without really testing any ideas? Of course it doesn’t. The alternative is to step back from individual hypotheses and to carry out what I call “targeted exploration”. This means that we do not feel it necessary to formulate our research in the simplistic “Ho, H1” format with a “yes/no” result structure. Instead, we take what information is available and try to identify patterns. If no information is available at all for some area, then we might explore it with the specific purpose of looking for patterns. Once a possible pattern is identified, we determine ways of testing how broadly it holds and what might be causing it. This involves more exploration, but specifically in areas that are intended to provide the necessary data to test the broad pattern. If the pattern holds, then we can formulate even more specific ideas about causation, leading eventually to the testing of particular hypotheses.
Some important points should be noted. First, targeted exploration does not conflict with focused hypothesis testing. Rather, it ultimately feeds into hypothesis-driven research, but is particularly important because it takes us into new territory rather than working within existing areas. Second, it is not done blind. There is a specific reason to target particular areas. Third, as it does not have a simple refuted/supported result but rather can be set up to reveal many different things, the results can be very informative either way. Finally, because it is based on large-scale sampling, exploration of this type has the beneficial side effect of closing some major gaps in our basic knowledge.
Let me give you an example of how this works.
Insects are by far the most diverse group of animals, at least in terms of described species. However, they have traditionally been poorly covered in animal genome size studies. When I was a graduate student, I compiled the Animal Genome Size Database, which made it possible to look across all the data that were available and see what patterns emerged. Based on work in amphibians, it was apparent that species with complex developmental programs including metamorphosis had smaller genomes than species without metamorphosis. I wondered if something similar might apply to insects, given that there are orders with complete metamorphosis (holometabolous development) and orders with incomplete metamorphosis (hemimetabolous development).
That is step 1: ask a question and look for a pattern. The data for insects were very limited, but it did seem as though insects with complete metamorphosis possess smaller genomes than those lacking complete metamorphosis, making this similar to the case in amphibians. However, there were not really enough data to say much about this, so as part of my graduate work I set out to get more insect data. I added a few hundred species, mostly just whatever I could get locally, and doing my best to include species from several orders with and without metamorphosis. That is step 2: assemble a dataset that can at least be used to identify a possible pattern. At this stage, the sampling is somewhat unconstrained — just get whatever you can, with the question still in mind. Why do it like this? Because a) you don’t have enough information to be very specific in what data you need, b) you’re working in a new area, so any data you get will be informative, and c) you don’t know if the pattern you are looking for is really the main pattern, so it is best to sample more widely in case some other pattern shows up.
Here is what I found:
 With the exception of one beetle species out of more than 150 (and I still want to check this myself), no insects with complete metamorphosis appear to have genome sizes larger than 2pg (~ 2 billion base pairs). On the other hand, orders without complete metamorphosis often include species with enormous genomes.
With the exception of one beetle species out of more than 150 (and I still want to check this myself), no insects with complete metamorphosis appear to have genome sizes larger than 2pg (~ 2 billion base pairs). On the other hand, orders without complete metamorphosis often include species with enormous genomes.
So, step 3 is then to see whether this holds with a broader sampling. Now we are getting into the targeted exploration. What we need is a) more data from holometabolous orders (do they exceed this threshold and we just haven’t found them?) and b) more from hemimetabolous orders (do most of them have examples that are larger than the threshold?). Since this possible pattern was identified, we have added hundreds of species from both kinds of insects, including about 400 butterflies and moths (holometabolous, none larger than 2pg), 90 wasps, ants, and bees (holometabolous, none larger than 2pg), 75 flies (holometabolous, none larger than 2pg), and 100 dragonflies (about 1/5 of known diversity in North America; hemimetabolous, a few larger than 2pg). So far, so good, and this work continues with current projects on wasps, flies, caddisflies, and stone flies. But questions remain: Does this hold in additional orders? Is there really a link between development and genome size in insects? Why 2pg? Are there other explanations (e.g., other constraints, phylogenetic effects, differences at the level of mutational mechanisms)?
For step 4, we started to test this idea that development constrains genome size in insects. First, we looked at the rate of development (egg to adult) within a single genus (Drosophila), and found a significant correlation with genome size. We have also started looking at “curious” orders that may be exceptions that prove the rule: for example, mayflies have an additional nymphal moult that other hemimetabolous orders don’t, so this may impose an additional constraint and keep their genomes small — I have only looked at one so far (yes, small), but I will let you know how it turns out once we do a large sample. We are also looking at specific comparisons within orders based on a combination of their traits (developmental rate, parasitic vs free living, body size, flight) and phylogenetic relationships. In this case, shifts in lifestyle are especially informative because they may illustrate an evolutionary association between genome size and the characteristics of interest.
Assuming these patterns hold up and we are convinced that development is linked with genome size, we will want to know how — thus, step 5. The most likely mechanistic bridge between genome size and organism development is cell division. However, no one has looked at cell division rate across insects with different genome sizes. This would be much more difficult than doing large-scale surveys, but it could be focused on a few representative species with different DNA amounts. If we really want to know if DNA content affects cell division, we would need to examine this experimentally in step 6 — for example, by actively adding or removing different amounts of DNA and observing the effects on cell cycle parameters. I have been trying for a few years to get funding to do this (in yeast initially), but no success.
I think it is obvious that this kind of approach falls outside the typical hypothesis-driven focus. However, it does get us from knowing almost nothing in step 1 to formulating and testing specific hypotheses in step 6. Along the way, we have greatly expanded the available dataset, and have revealed several additional patterns worh exploring within some orders. If I had to express each step in the form of hypotheses, I probably could, but because we are exploring so many questions at once in each step, it makes more sense to just think about questions and make sure the sampling will allow us to generate answers. Without the existing knowledge base, focusing on one hypothesis only is premature and very limiting in what it will accomplish.
Obviously, we are not just interested in insects. Over the rest of the series, I will talk about other groups that we are eager to explore, and will discuss in more detail some of the focused work on mechanisms that I am interested in. Some of these therefore begin at step 1, others at step 6, and some somewhere in between.
What would I do with more research support? Part One: Background.
One of the great joys of being a scientist is that we get to spend our lives exploring the aspects of the natural world that most intrigue and excite us. However, the equally great frustration of being a researcher is that our curiosity and passion invariably outstrip the resources available for our explorations. It often feels like we spend the bulk of our creative energy begging for money, and when this is declined — as it often is — it can be crushing. What keeps us going is the conviction that what we are doing, and what we have not yet found a way to do, is interesting and important and worth pursuing.
The primary focus of my research is the evolution of genome size in animals. Genome size is the amount of DNA in one copy of the chromosome set of a species, generally measured in terms of the number of base pairs (bp) or in mass (in picograms, or 10-12g). What makes this an intriguing topic of research is the enormous variability that exists across species: in animals, genome sizes range more than 7,000-fold. Think about that for a moment. Some animals have 7,000 times more DNA in their cells than others. Even within vertebrates, there is huge diversity at the genomic level: the largest (lungfish) is 350 times larger than the smallest (pufferfish). Or consider amphibians, which range about 120-fold from the smallest in some frogs to the largest in a few aquatic salamanders.
The human genome contains about 3.2 billion base pairs. In the simplest terms, one might expect this to be the largest genome of all — humans are the most complicated organisms (right?) and that should require the most genes (right?) which in turn means more DNA (right?). This was indeed the assumption when researchers began assessing genome sizes in the late 1940s — before the structure of DNA was elucidated, and even before it had been established that DNA is the hereditary molecule. At this time it was reported that the amount of DNA in a species’ cells is mostly constant (thus, genome size is also called “C-value”). This itself was suggested to indicate that DNA, and not protein, serves as the molecular basis of inheritance. However, it was also obvious by 1951 that the amount of DNA varies dramatically among species, and that the “complexity” of an animal and its genome size are decoupled. There are, it was discovered, salamanders with 40x more DNA per genome than in humans. This made no sense. DNA amount is constant within species because it is what genes are made of, and yet more complicated organisms (which presumably require more genes) may have substantially less DNA in their genomes than simpler organisms. This became known as the “C-value paradox” in the early 1970s.
It was not long before the apparent “paradox” was resolved: most DNA in animal and plant genomes is not genes (it is “non-coding DNA”). This means that genome size need not be related to the number of protein-coding genes, and that there is no reason to expect more complex animals to have more DNA in their genomes. However, this raised many new questions: What is this non-coding DNA? Where does it come from? How does it increase or decrease in amount in different genomes? Does it have any effect on the organism? Does it have any function? Why do some species have so much of it and others so little?
Despite several decades of research, most of these questions remain at best only partially answered. This is where my lab’s research comes in. We are interested in genome size diversity across all animals, in its effects on organism biology, and in the factors ranging in scale from individual DNA elements to ecological properties that accentuate or constrain amounts of DNA in the genomes of different species.
One thing that has become clear over the past several decades is that genome size is not randomly distributed across taxa. Some, like birds, all seem to have relatively small genomes. Others, like salamanders, all have large genomes. The quantity of DNA also relates to important features such as cell size and cell division rate, such that large genomes are found in cells that are big and divide slowly. Because all animals are made of cells, this means that any feature relating to cell size or cell division rate could be indirectly related to genome size. Body size is an obvious possibility, at least when cell numbers are held mostly constant. Metabolic rate is another possibility, because the larger a cell gets, the lower its relative surface area is, and this can influence gas exchange. Developmental rate is yet another, because slower individual cell divisions can add up to protracted development overall.
We have found that body size is correlated with genome size not only in some invertebrates like flatworms and copepod crustaceans, but also within specific groups of vertebrates like rodents, bats, and birds. Inverse relationships between genome size and metabolic rate have been reported in both mammals and birds, and in particular it has been argued that flight imposes a constraint on genome size due to its high metabolic demands. This latter idea has been around for several years, but it has recently become the subject of renewed interest and some intriguing new discoveries. For example, my colleague Chris Organ has used fossil cell size measurements to reveal that theropod dinosaurs (the lineage from which birds evolved) already had somewhat reduced genome sizes relative to other lineages before birds evolved, and that pterosaurs (the first vertebrates to evolve flight) also had small genomes. One of my students has been working on flight in birds, and showed that wing parameters associated with flight ability are related to genome size as well. We have also found recently that hummingbirds have the smallest genomes among birds (this isn’t published yet, but we’re writing the paper as we speak).
In terms of development, we have found in insects like lady beetles and vinegar flies that larger genomes are associated with slower overall development. Similar correlations have been known for some time in amphibians. What is more interesting is the pattern that we see with regard to metamorphosis, which represents a period of rapid and extreme physical reorganization. Groups with intensive metamorphosis, like frogs living in deserts that complete their life cycle quickly during wet seasons, have very small genomes (smaller than birds). Others, like aquatic salamanders that have lost the ability to metamorphose, have some of the largest genomes among animals. This also seems to apply to the major lineages of insects. Orders exhibiting complete metamorphosis (“holometabolous development”) appear almost never to exceed about 2 billion base pairs, whereas some without complete metamorphosis (“hemimetabolous development”) can be very large — there are grasshoppers with 5x more DNA than in humans.
Although genome size has been investigated for more than 60 years, some of these trends are only now coming to light. One reason is that we are focusing on the “big picture” now. Another reason is that we have technology that allows us to estimate genome sizes for large numbers of species. To give one example, an undergraduate student and I produced new data for more than 300 species of moths last summer alone. Previously, only 50 moth species had been analyzed (almost all of them in a pilot study I did a few years ago). Of course, this is a miniscule fraction of the 180,000 or so described species in the order, but it’s infinitely better than no information at all. Various students of mine have begun filling other major gaps, including in mammals, birds, insects, worms, and molluscs, but a huge amount of work remains just to get a basic picture of genomic diversity and its significance.
Over the upcoming series of posts, I will highlight some of the projects that I am very interested in undertaking, but which are on indefinite hold due to lack of funds. (It’s not that I haven’t tried — but granting agencies tend not to like this kind of large-scale “discovery” science as compared to the testing of very focused hypotheses). There are several reasons why I think it is worth doing this. First, most members of the public get only snippets of what goes on in research labs, most often provided by news reports. The raw curiosity that drives basic research is not often conveyed, particularly when projects are first conceived (vs. once they’re completed and published). Second, this is the stuff that gets me out of bed in the morning, and I hope that others can share in the excitement that my students and I feel when we think about, and try to answer, these fundamental questions about the diversity of life. Third, I believe it is useful for people to grasp the frustration that every scientist lives with when he or she feels that there are great ideas collecting dust for simple lack of funds. Finally, it provides an opportunity to talk about some intriguing animal groups from a perspective that most people haven’t considered. In that sense, it should be an interesting exercise in thinking about the wondrous biological diversity that surrounds us.
In the meantime, you are welcome to explore the Animal Genome Size Database to get a sense of the tremendous diversity — and glaring gaps in our knowledge — that drive my research program.
Recent lab papers.
So, what have we been up to in the lab?
Ardila-Garcia, A.M. and T.R. Gregory (2009). An exploration of genome size diversity in dragonflies and damselflies (Insecta: Odonata). Journal of Zoology, in press.
Smith, J.D.L. and T.R. Gregory (2009). The genome sizes of megabats (Chiroptera: Pteropodidae) are remarkably constrained. Biology Letters, in press.
Smith, E.M. and T.R. Gregory (2009). Patterns of genome size diversity in the ray-finned fishes. Hydrobiologia 625: 1-25.
Andrews, C.B. and T.R. Gregory (2009). Genome size is inversely correlated with relative brain size in parrots and cockatoos. Genome 52: 261-267.
Andrews, C.B., S.A. Mackenzie, and T.R. Gregory (2009). Genome size and wing parameters in passerine birds. Proceedings of the Royal Society of London B 276: 55-61.
Currently finishing up:
Ants/bees/wasps, butterflies, moths, hummingbirds, more bats, more birds
Speaking of small genomes…
… our paper on megabats was published online yesterday. It’s free to access at the moment. Turns out megas have even more constrained genomes than microbats.
Abstract
It has long been recognized that bats and birds contain less DNA in their genomes than their non-flying relatives. It has been suggested that this relates to the high metabolic demands of powered flight, a notion that is supported by the fact that pterosaurs also appear to have exhibited small genomes. Given the long-standing interest in this question, it is surprising that almost no data have been presented regarding genome size diversity among megabats (family Pteropodidae). The present study provides genome size estimates for 43 species of megabats in an effort to fill this gap and to test the hypothesis that all bats, and not just microbats, possess small genomes. Intriguingly, megabats appear to be even more constrained in terms of genome size than the members of other bat families.
With genomes, bigger may really be better…
…as targets for genome sequencing in order to avoid bias in what we discover about gene regulation from sequenced genomes, because so far only small genomes have been sequenced. Such is the message reported at the HHMI based on a recent paper by Michael Eisen. I have written about the major problem of drawing broad conclusions from the biased sequenced genome dataset, and I am very excited to see someone else noting that we really need to examine more diversity. I have been meaning to write a paper on why we need large-scale genome size surveys and why sequencing people should be enthusiastic about it (maybe even help fund it). Here is another great reason that I will cite.
It so happens that a student in my lab will soon be initiating a project on dipteran genome sizes — this gives it even broader significance. I might point out that tephritids do not have “big genomes” for insects by my reckoning (for that, you would need to get beyond holometabolous orders). Finally, if you’re wondering why Drosophila genomes are so streamlined, it actually looks like development may constrain how large they can be.
Bloodlust.
Depending on the animals in question, the amount of DNA per cell may be associated with body size, metabolic rate, developmental rate, or other traits. With an old fashioned cytogenetic staining method (the Feulgen reaction) and a new image analysis densitometry setup, we can estimate genome size for vertebrate species quite readily with only an air-dried sample of blood cells on a microscope slide. Getting the blood is the limiting step in many instances — in particular from cool and recently discovered critters like these that are now officially on my blood smear wish list.
- Paedocypris spp. – Fishes considered the world’s smallest vertebrates.
- Schindleria brevipinguis – Another tiny fish.
- Furcifer labordi — A reptile with the fastest life cycle of any terrestrial vertebrate.
- Barbourula kalimantanensis — The first lungless frog known.
- Nototriton sp. — A newly discovered miniature salamander.
- Leptotyphlops carlae — Newly discovered miniaturized snake.
If you taxonomists out there wouldn’t mind making smears for me when you find these kinds of beasts, that would be excellent.
Small genome sizes in pterosaurs, too.
My colleagues Chris Organ and Andrew Shedlock, who provided evidence that theropod dinosaurs already had (somewhat) reduced genome sizes prior to the evolution of birds (Organ et al. 2007) have followed up their study by estimating the genome sizes of several species of pterosaurs.
Pterosaurs were the first vertebrates to evolve powered flight, having taken to the air 70 million years before birds and 150 million years prior to bats. Interestingly (though perhaps not surprisingly at this point), they seem to have possessed reduced genome sizes, and these downsizings of DNA amount began before flight arose.
On the other hand, it is clear that the estimates for non-avian dinosaurs are not as small as modern birds and that the estimated ancestral genome size for birds was larger than the genome seen in various groups. Patterns can be observed in terms of flight ability across living avian species. Notably, my student Chandler Andrews showed that genome size is correlated with wing loading (and indication of flight capacity) within perching birds, and we are currently writing up major projects on bird groups with different flight ability as well as a study of hummingbirds; Jill Smith, another student, also has a large bat study to write up.
The story thus seems to be that genome reduction occurred in the dinosaur lineage of which birds are descendants before flight (so did feathers, bipedalism, and other characteristics), but were later further adjusted when flight arose (as were feathers, etc.). The same reductions before flight probably occurred in the pterosaur and bat ancestors. So it’s not flight per se that matters, but a feature linked with flight.
As Organ and Shedlock put it, “we hypothesize that a metabolic intensity required for flight, not flight itself, explains the correlated evolution between genome size and flight in amniotes.” — this seems very plausible given the growing amount of data on this topic.
References
Andrews, C.B., S.A. Mackenzie, and T.R. Gregory. Genome size and wing parameters in passerine birds. Proceedings of the Royal Society of London B, in press.
Organ, C.L., A.M. Shedlock, A. Meade, M. Pagel, and S.V. Edwards. 2007. Origin of avian genome size and structure in non-avian dinosaurs. Nature 446: 180-184.
Organ, C.L. and A.M. Shedlock. 2008. Palaeogenomics of pterosaurs and the evolution of small genome size in flying vertebrates. Biology Letters, in press.
Zimmer, C. 2007. Jurassic genome. Science 315: 1358-1359.
Gecko genome size and cell size.
One of the many aggravations I encounter when reviewing manuscripts is that some authors greatly overstate the applicability of statistically significant patterns they report. For example, a statistically significant pattern in a small comparison of a few animals may be extrapolated in the discussion to the kingdom at large.
Today I was disappointed to see a paper that is soon to come out in Zoology that does the opposite — i.e. takes a non-significant relationship in a handful of species and pretends that it challenges the importance of broad relationships that have been considered important for decades.
The paper in question is:
Starostova, Z., L. Kratochvil, and M. Flajshans. 2008. Cell size does not always correspond to genome size: phylogenetic analysis in geckos questions optimal DNA theories of genome size evolution. Zoology, in press.
They compared genome size and cell size across 15 geckos and found no correlation. From this, they went on to argue that genome size does not causally influence cell size and that genome size is not under selection due to cell size impacts.
First, let me point out that strong, positive correlations between genome size and cell size have been reported within and across all vertebrate classes including reptiles. So, on a broad scale, the relationship is clear.
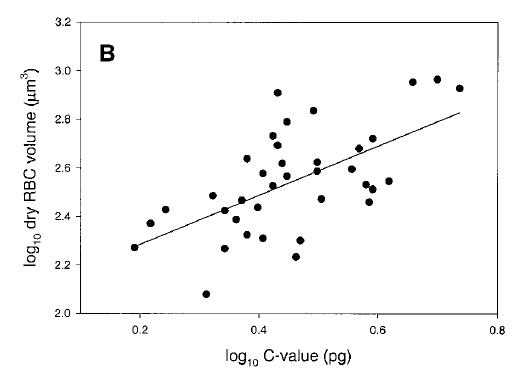
Genome size and cell size in reptiles. From Gregory (2001), based on data from Olmo and Odierna (1982).
Second, let me say that I have issues with their methods. For example, they used DAPI as the fluorochrome, which is base-pair specific and can give biased determinations (they recognize this but assume the species are all the same in AT content). Second, they produced fairly substantial error ranges in their measurements given that these were all raised in the lab or obtained from pet shops and not taken from different wild populations (i.e., the variability between conspecifics is probably artifact). Third, they counted “forms” of the same species from different places as being independent in their analyses — so it wasn’t 15 species, rather it was 12 species with several represented by multiple points.
These are not the main problems, though. The first is that they clearly had outliers in the dataset. In particular, Coleomyx brevis (CB) and Coleomyx variegatus (CV) have “large” (~2pg) genomes but comparatively small cells. I don’t think I even need to draw the line through the remaining points, but in case eyeball statistics don’t do it, the correlation is highly significant without them (r = 0.74, p < 0.006) (they recognize this, too, but note the title they chose for the paper nonetheless).
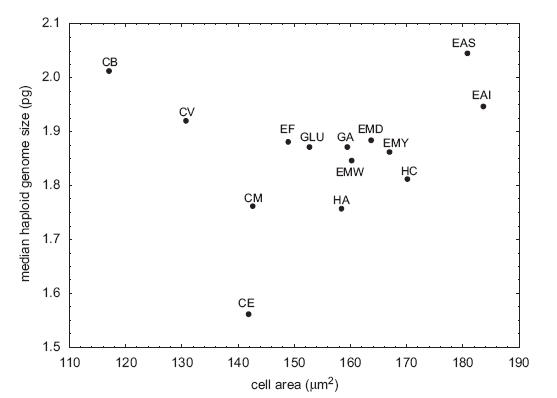
From Starostova et al. (2008).
So, how can this be explained? Well, you have to know something about nucleotypic theory, which these authors actually did mention. It’s not genome size all alone that is the determining factor — nucleus size is critical. The “nucleotype” is defined as “that condition of the nucleus that affects the phenotype independently of the informational content of the DNA†(Bennett 1971). As has been pointed out repeatedly (e.g., by me, Cavalier-Smith, Bennett, and others), the compaction level of DNA in the nucleus adds a second dimension to the relationship. More DNA is one thing, but if it is compressed into a tightly packed, reduced nucleus, then cell size may still be small.
That leads to the second major problem. Looking at the data reported in a previous study (Starostova et al 2005), there is no correlation between genome size and nucleus size. There is, however, a positive correlation between nucleus size and cell size across these reptiles.
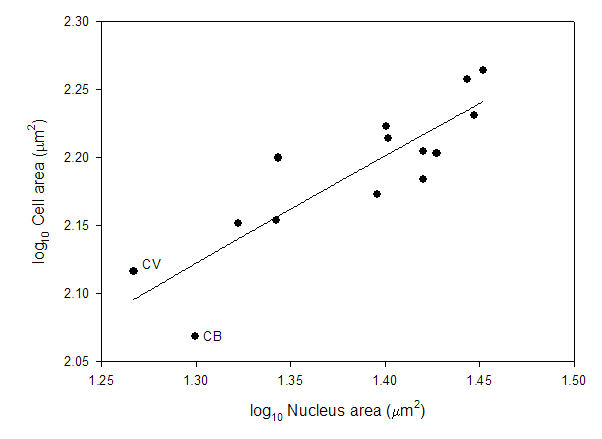
Based on databy Starostova et al. (2005).
The two outliers in the genome size vs cell size comparison have more compact nuclei and this allows smaller cell sizes with larger genome size. Cell size is correlated with body size in these geckos, and these two species are “dwarfs” (~4.5g) relative to other species (as big as ~90g). So, there could very easily be selection for reduced cell size which, in this narrow range in DNA amount, was met by a compaction of the nucleus rather than a loss of DNA.
This actually reinforces the strength of nucleotypic theory.
______
Bennett, M.D. 1971. The duration of meiosis. Proceedings of the Royal Society of London B 178: 277-299.
Gregory, T.R. 2001. The bigger the C-value, the larger the cell: genome size and red blood cell size in vertebrates. Blood Cells, Molecules, and Diseases 27: 830-843.
Olmo, E. and G. Odierna. 1982. Relationships between DNA content and cell morphometric parameters in reptiles. Basic and Applied Histochemistry 26: 27-34.
Starostova, Z., L. Kratchovil, and D. Frynta. 2005. Dwarf and giant geckos from the cellular perspective: the bigger the animal, the bigger its erythrocytes? Functional Ecology 19: 744-749.
Genome size and complexity (again).
Some time ago, I wrote about the (non-)relationship between genome size and gene number, which also included some discussion of the obvious decoupling of DNA content and “morphological complexity” (however defined). Now, Steve Matheson of Quintessence of Dust has a fun way of demonstrating this, by asking readers to guess which animals have larger genomes than which others based on intuitive concepts of complexity. Even I have to think about it for some of them, and I actually measured some of those genome sizes! (In part, this is because there is often significant diversity in genome size within groups of morphologically similar species, which in itself shows the disconnect between complexity and DNA amount). The first two installments of the quiz are here and here. Have fun.